Metabolic & Genetic Disorders
Focused Care for Diabetes, Hyperglycemia, Urea Cycle Defects, and Genetic Liver Disorders through Advanced Molecular Insights
Metabolic and genetic disorders represent a complex interplay of biochemical dysfunctions and inherited anomalies that significantly impact patient health. At CLRD, our treatment specialization is rooted in precision medicine, leveraging advanced molecular insights to deliver targeted interventions for conditions such as diabetes, hyperglycemia, urea cycle defects, and genetic liver disorders.
Advanced Care through Molecular Insights
Metabolic and genetic disorders are chronic, often progressive conditions that disrupt core biochemical pathways and place sustained stress on the liver’s detoxification and synthetic functions. At CLRD, treatment specialization is built on precision diagnostics and regenerative therapies that directly correct cellular deficits rather than merely compensating for them. Our programs integrate molecular genetics, enzyme physiology, cell therapy, and organ bioengineering to deliver durable outcomes for diabetes and hyperglycemia, urea cycle defects, and inherited liver disorders.
Diabetes and Hyperglycemia
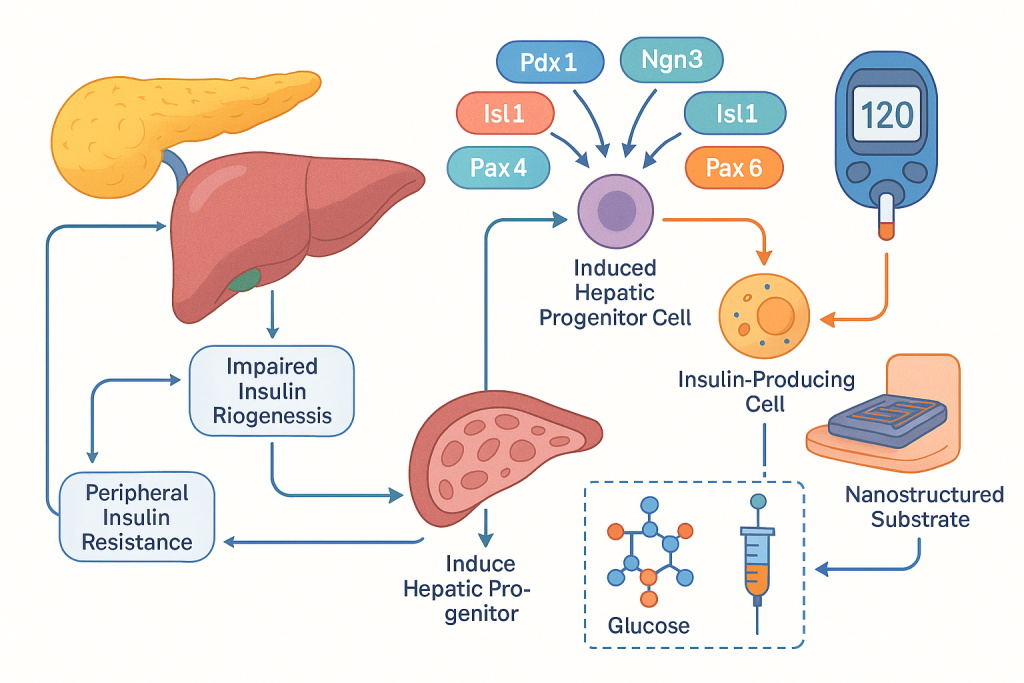
Diabetes and hyperglycemia management at CLRD begins with molecular stratification of each patient’s disease biology, including assessment of pancreatic transcription factor networks and hepatic glucose handling to determine whether impaired insulin biogenesis, peripheral resistance, or aberrant hepatic gluconeogenesis is dominant. Therapeutic planning pairs pharmacogenomics-guided agents with regenerative strategies that restore endogenous glycemic control. Induced hepatic progenitor cells are directed toward insulin-producing phenotypes by modulating Pdx-1, Ngn-3, Isl-1, Pax-4, Pax-6 and Nkx-6.1 expression, enabling autologous or allogeneic cell therapy that augments physiologic insulin release. This framework is supported by device and materials science, where glycemic stimulation on conducting nanostructured substrates has been shown to enhance insulin secretion and reverse hyperglycemia in preclinical models, informing our translational pipeline for cell–device assisted glycemic restoration. Longitudinal follow up uses continuous glucose analytics and metabolic biomarker panels to verify durability and safety of the restored glycemic set point while tapering exogenous insulin as endogenous production stabilizes.
Urea Cycle Defects
Urea cycle defects present as hyperammonemia with risk of cerebral edema and neurologic compromise, requiring immediate ammonia detoxification alongside definitive enzyme correction. CLRD’s pathway-focused treatment model combines rapid nitrogen load reduction with molecular and cellular restoration of ureagenesis. Diagnostic work-up includes genetic delineation of the affected enzyme and functional assays of urea cycle activity to quantify residual capacity. Therapeutic escalation employs ammonia-scavenging pharmacology and targeted nutrition while concurrently deploying hepatocyte or progenitor cell transplantation to reinstate hepatic ammonia detoxification. Our clinical and translational experience with microencapsulated hepatocytes and xenogenic models has established the feasibility of immune-protected cell sources when autologous tissue is unavailable, and bioengineered grafts are used as a bridge to definitive cellular correction. Long-term care focuses on maintaining metabolic stability, preventing catabolic crises, and safeguarding neurocognition with structured rehabilitation as ammonia handling normalizes.
Genetic Liver Disorders
Inherited liver disorders require therapy that addresses the causal molecular lesion. CLRD designs patient-specific corrective plans that combine gene-level diagnosis, cellular replacement of deficient functions, and organ support during acute decompensation. In bilirubin conjugation defects such as Crigler–Najjar syndrome, hepatic progenitor cell infusion has been used to lower unconjugated bilirubin and reduce phototherapy dependence, effectively postponing or eliminating the need for transplant in selected cases. In acute liver failure and cholestatic conditions including biliary atresia, fetal hepatocyte and hepatic progenitor infusions have provided immediate metabolic support, while bioartificial liver systems supply temporary detoxification and synthetic capacity until cellular integration is sufficient. For protein misfolding and storage disorders such as alpha‑1 antitrypsin deficiency and Wilson’s disease, CLRD adopts a staged approach where molecular chaperone modulation and copper chelation are paired with regenerative replacement of functional hepatocyte mass, moving patients from disease control toward molecular remission.
Real‑World Case Studies
Crigler–Najjar Syndrome with Refractory Hyperbilirubinemia
A pediatric patient with genetically confirmed Crigler–Najjar type I presented with escalating unconjugated bilirubin and recurrent phototherapy admissions. CLRD implemented hepatic progenitor cell transplantation through the hepatic artery following characterization of the patient’s conjugating enzyme deficit. Post‑infusion monitoring showed a marked and sustained reduction in serum bilirubin, decreased phototherapy requirements, and improved neurodevelopmental trajectories over twelve months. The cell therapy postponed consideration of whole‑organ transplant and provided a window for growth and stabilization while a longer‑term corrective plan was finalized.
Fulminant Hepatic Failure Bridged by Fetal Hepatocyte Transplantation
An adult with fulminant hepatic failure underwent urgent molecular and metabolic profiling at CLRD. Human fetal hepatocyte transplantation was performed to provide rapid biochemical support. Within days, ammonia levels, coagulopathy indices, and lactate normalized sufficiently to allow recovery of native liver function. The patient was discharged with normalized transaminases and remained transplant‑free at one‑year follow‑up, highlighting the utility of cellular bridging therapy in acute decompensation.
Acute Fatty Liver of Pregnancy Managed with Hepatocyte Support
A third‑trimester patient presented with acute liver failure due to acute fatty liver of pregnancy, complicated by coagulopathy and encephalopathy. CLRD deployed peritoneal transplantation of human fetal hepatocytes as metabolic support to augment detoxification and ureagenesis while obstetric and intensive care teams managed maternal–fetal stabilization. The cellular support improved ammonia clearance and synthetic function, facilitating safe delivery and maternal recovery without the need for emergent liver transplant, and liver indices normalized in the postpartum period.
Urea Cycle Defect Stabilized with Microencapsulated Hepatocytes
An adolescent with recurrent hyperammonemic crises due to a urea cycle enzyme mutation underwent CLRD therapy using microencapsulated hepatocytes to reconstitute ammonia detoxification while minimizing alloimmune responses. Following implantation, ammonia levels remained within safe thresholds through catabolic stress episodes, hospitalizations decreased, and neurocognitive testing showed stabilization. Nutritional and pharmacologic strategies were gradually de‑intensified as cellular function persisted, demonstrating a durable metabolic correction without full organ transplant.
CLRD Capability in Diagnosis and Treatment
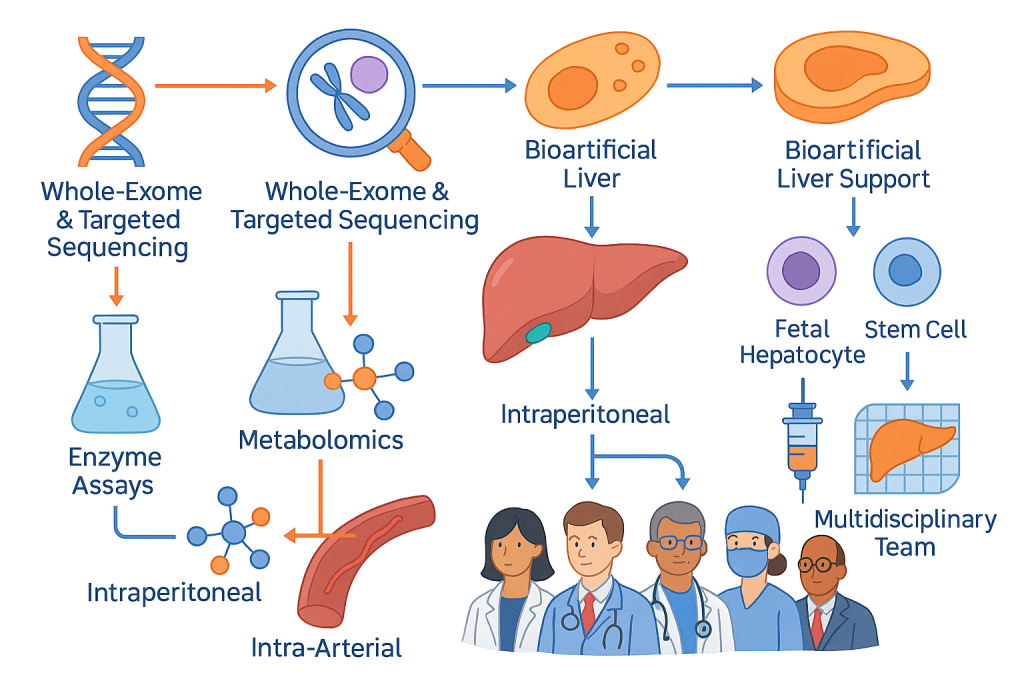
CLRD’s capability spans comprehensive molecular diagnostics and advanced therapy delivery. Whole‑exome and targeted gene sequencing define pathogenic variants and guide therapy selection. Enzyme assays and metabolomics quantify residual pathway capacity and track therapeutic response. Cellular therapies include fetal hepatocytes, hepatic progenitors, and carefully characterized stem cell derivatives administered via intraperitoneal or intra‑arterial routes to maximize engraftment and metabolic rescue. Organ bioengineering capabilities provide bioartificial liver support and humanized liver grafts for acute stabilization and chronic supplementation, and microencapsulation technology adds an immune‑protective layer that broadens donor options. Care pathways are embedded in multidisciplinary teams, with critical care, endocrinology, hepatology, maternal–fetal medicine, and rehabilitation aligned to deliver seamless molecularly informed treatment from crisis through convalescence.
Future Directions
Future directions at CLRD focus on scaling precision cell therapies and integrating gene correction to achieve definitive cures. Gene‑edited hepatic progenitors are being advanced for enzyme replacement in bilirubin conjugation defects, copper transport disorders, and protease inhibitor deficiencies, allowing correction at the source cell before transplantation. Humanized bioengineered livers are being refined to serve as off‑the‑shelf metabolic support platforms that can be deployed rapidly in acute failure, reduce waiting time for donor organs, and provide a safe bridge while native liver regeneration proceeds. For diabetes, hybrid cell–device systems are moving toward outpatient deployment, where trans‑differentiated insulin‑producing cells interface with smart conductive matrices to deliver physiologic pulsatile insulin independent of systemic immunosuppression. In urea cycle disorders, next‑generation microencapsulation materials and immune‑stealth coatings are being validated to extend graft longevity and resilience during catabolic stress, further reducing crisis admissions. Longitudinal molecular monitoring using circulating cell‑free DNA and RNA, mitochondrial biomarkers, and microRNA panels is being incorporated into routine care to predict decompensation before clinical onset, enabling preemptive therapy adjustments that maintain biochemical stability. Across programs, CLRD is standardizing manufacturing and quality systems for clinical‑grade cells and bioengineered grafts, expanding access while preserving the safety and efficacy demonstrated in our translational work.